

La filogenética es clave para descubrir la historia evolutiva desconocida.
Todas las especies que habitan en el planeta Tierra tienen un antepasado común. La evolución fue dando lugar a diferentes especies, siguiendo caminos a veces misteriosos. Aunque la historia evolutiva de algunos grupos de especies es conocida, quedan por determinar las relaciones de miles de ellas. El genoma es una de las herramientas empleadas para enfrentar este reto, en un campo llamado filogenética, en el que colaboran biólogos, estadísticos, matemáticos e informáticos.
El objetivo final de esta disciplina sería poder describir todas las relaciones entre cualquier conjunto concreto de especies, a través de un esquema llamado árbol filogenético. En este diagrama las hojas representan las especies actuales; la raíz, el ancestro común a todas esas especies; los nodos, las especies ancestrales; y la división de ramas, la creación de especies. Por ejemplo, al considerar el gorila, el chimpancé y el humano, hay sólo tres árboles filogenéticos que podrían explicar su evolución: el que presenta al gorila y al chimpancé como especies más cercanas (con un ancestro común), el que une al gorila y humano y el que junta chimpancé y humano, como muestra la figura.
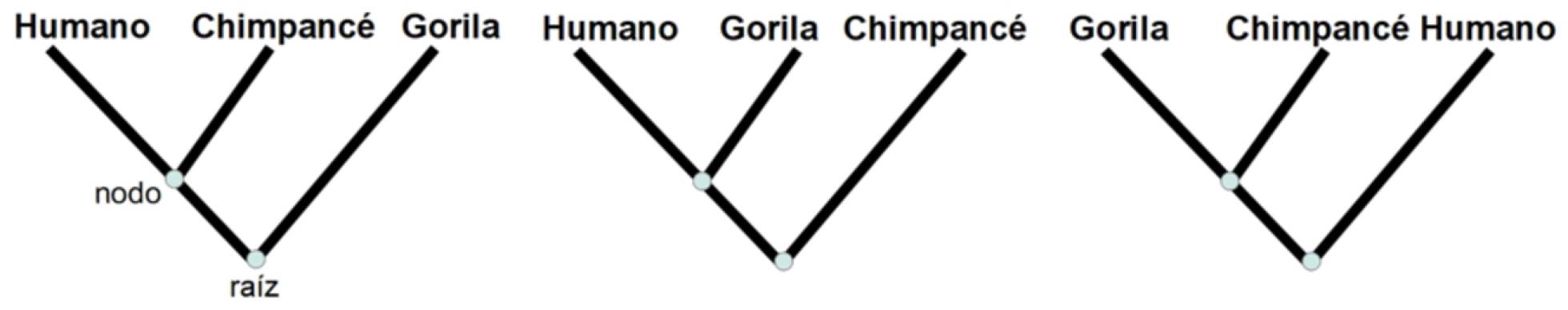
Ejemplo de árbol filogenético de gorila, humano y chimpancé MARTA CASANELLAS
A partir del genoma de las especies actuales se intenta deducir qué configuración, entre todos los posibles árboles, parece la más apropiada. Como no disponemos del genoma de las especies ancestrales, se emplean modelos de evolución de las secuencias de ADN a lo largo del árbol. Los parámetros de estos modelos, que desconocemos, son las probabilidades de mutación de las secuencias a lo largo del proceso evolutivo en cada rama. A partir de los datos genómicos de las especies actuales, se escoge el árbol y los parámetros que maximizan la probabilidad de observar los genomas de las especies actuales. Este proceso de optimización necesita gran cantidad de datos (secuencias de ADN muy largas); como esto no siempre es posible, la inferencia está sujeta a error.
Pero, además, este método solo se puede emplear para estudiar grupos de menos de veinte especies, ya que el número de árboles filogenéticos va aumentando de forma exponencial según el número de especies a estudiar. Para tres, sólo hay tres posibles configuraciones, como ya hemos visto. Pero cuando se añade una nueva especie, se obtiene un total de quince árboles filogenéticos posibles, y para 50 especies, se obtienen alrededor de 3x1076 posibilidades (este número no está muy lejos del número estimado de partículas del universo). Este crecimiento de las opciones hace inviable inferir el árbol comprobando todos los árboles posibles. Una forma de inferir el árbol de un grupo grande de especies es usar la aglomeración de árboles más pequeños. Este método tiene sus problemas, ya que las piezas pequeñas pueden ser incompatibles entre ellas. Alternativamente, también se puede construir directamente un árbol estimando la distancia evolutiva entre las especies, es decir, estimando la cantidad de mutaciones que han sufrido durante la evolución. A partir de aquí se escoge el árbol que minimiza la distancia evolutiva total entre las especies del árbol.
Recientemente han surgido técnicas que se enmarcan dentro de una nueva disciplina llamada estadística algebraica. Estas herramientas están basadas en el estudio algebraico de los modelos de evolución y permitirían la inferencia del árbol filogenético sin tener que estimar las probabilidades de mutación del ADN. Estos métodos están en sus primeros pasos de desarrollo, son válidos de momento para reconstruir árboles con un número pequeño de especies y hay que adaptarlos a los métodos de aglomeración para poder construir árboles mayores y usarlos con datos reales del genoma. Pero hasta ahora han dado buenos resultados en datos simulados y ya se han implementado en alguno de los paquetes de software más usados por los biólogos.
Todos estos esfuerzos en el avance de la filogenética no sólo son útiles para entender la historia evolutiva y, por ejemplo, determinar en qué momento se separaron dos grupos de especies, sino que las mismas herramientas se emplean también en genómica (para determinar elementos funcionales del genoma), en biomedicina (por ejemplo, para determinar el origen de cepas de virus) e incluso en lingüística.
Marta Casanellas es profesora del Departament de Matemàtiques de la Universitat Politècnica de Catalunya